SeqFindR
A tool to easily create informative genomic feature plots
SeqFindR
SeqFindR - easily create informative genomic feature plots.
This is an early release version of SeqFindR. The tool is still undergoing rapid development. We have only tested SeqFindR on linux systems.
Requirements
You'll need to install/have installed:
- ncbiblast >= 2.2.27
- python >= 2.7 (not Python 3 supported)
We also use the following python libraries:
- numpy >= 1.6.1
- scipy >= 0.10.1
- matplotlib >= 1.1.0
These should be installed automatically If you follow the instructions below.
Installation
You'll need to have git installed. As a scientist git can be really useful. See here for some discussion.
Option 1 (with root/admin):
cd ~/
git clone git://github.com/mscook/SeqFindR.git
cd SeqFindR
sudo python setup.py install
Option 2 (standard user) replacing INSTALL/HERE with appropriate:
cd ~/
git clone git://github.com/mscook/SeqFindR.git
cd SeqFindR
echo 'export PYTHONPATH=$PYTHONPATH:~/INSTALL/HERE/lib/python2.7/site-packages' >> ~/.bashrc
echo 'export PATH=$PATH:~/INSTALL/HERE/bin' >> ~/.bashrc
source ~/.bashrc
python setup.py install --prefix=~/INSTALL/HERE/SeqFindR/
If the install went correctly:
user@host:~/> which SeqFindR /INSTALL/HERE/bin/SeqFindR user@host:~/> SeqFindR -h
Please regularly check back or git pull & python setup.py install to make sure you're running the most recent SeqFindR version.
Example figure produced by SeqFindR
SeqFindR CU fimbriae genes image. 110 E. coli strains were investigated. Order is according to phylogenetic analysis.

SeqFindR database files
The SeqFindR database is in multi-fasta format. The header needs to be formatted with 4 comma separated elements.
The elements are:
- identifier,
- common name,
- description and
- species
The final element, separated by [] contains a classification.
An example:
>70-tem8674, bla-TEM, Beta-lactams Antibiotic resistance (ampicillin), Unknown sp. [Beta-lactams]
AAAGTTCTGCTATGTGGCGCGGTATTATCCCGTGTTGACGCCGGGCAAGAGCAACTCGGTCGCCGCATAC
>70-shv86, bla-SHV, Beta-lactams Antibiotic resistance (ampicillin), Unknown sp. [Beta-lactams]
CTCAAGCGGCTGCGGGCTGGCGTGTACCGCCAGCGGCAGGGTGGCTAACAGGGAGATAATACACAGGCGA
>70-oxa(1)256, bla-OXA-1, Beta-lactams Antibiotic resistance (ampicillin), Unknown sp. [Beta-lactams]
>70-tetB190, tet(B), Tetracycline Antibiotic resistance (tetracycline), Unknown sp. [Tetracycline]
CAAAGTGGTTAGCGATATCTTCCGAAGCAATAAATTCACGTAATAACGTTGGCAAGACTGGCATGATAAG
Tutorial
Navigate to the SeqFindR/example directory (from git clone). The following files should be present:
- A database file called Antibiotic_markers.fa (-d option)
- A ordering file called dummy.order (-i option)
- An assemblies directory containing strain1.fa, strain2.fa and strain3.fa (-a option)
- A consensus directory containing strain1.fa, strain2.fa and strain3.fa (-m option)
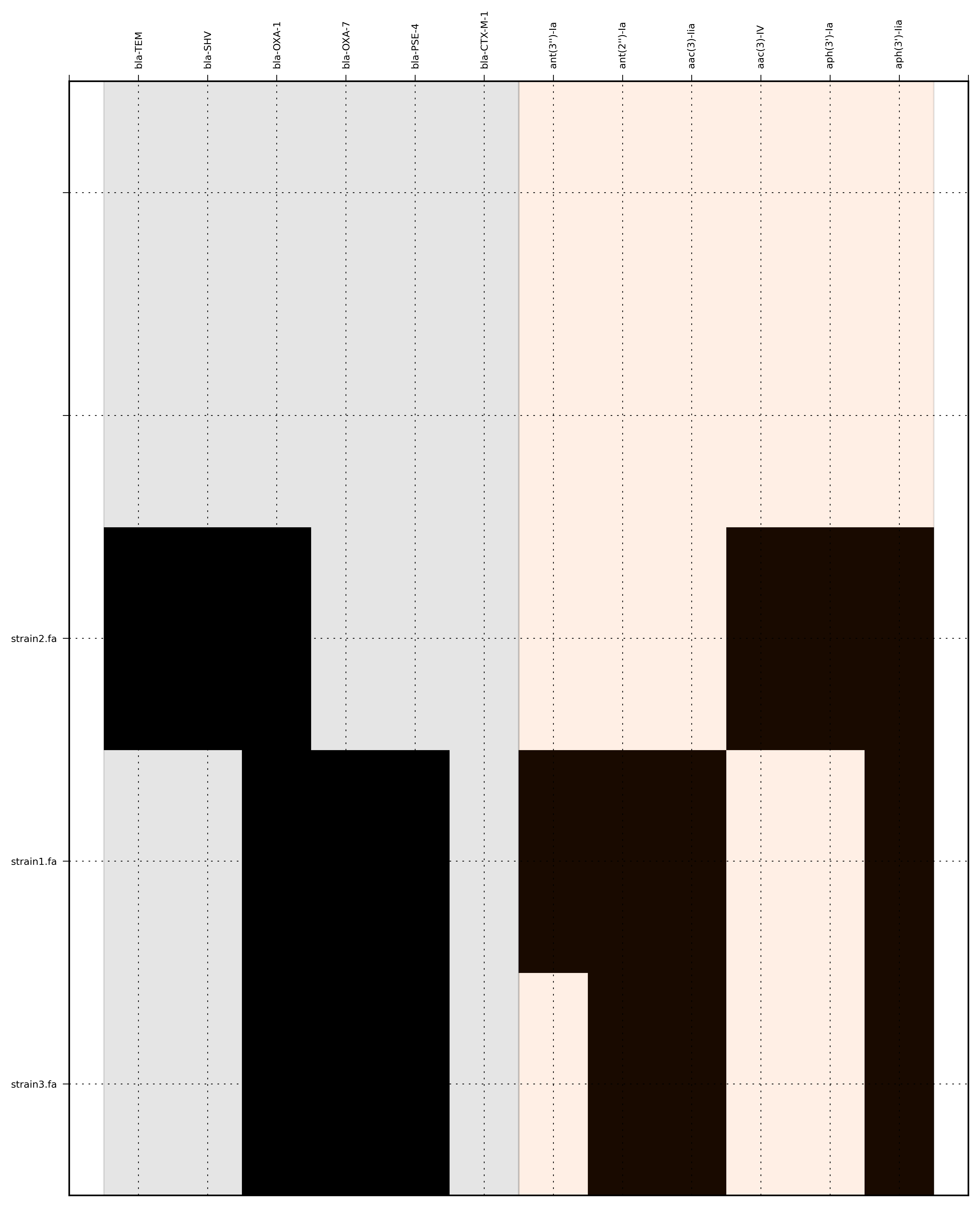
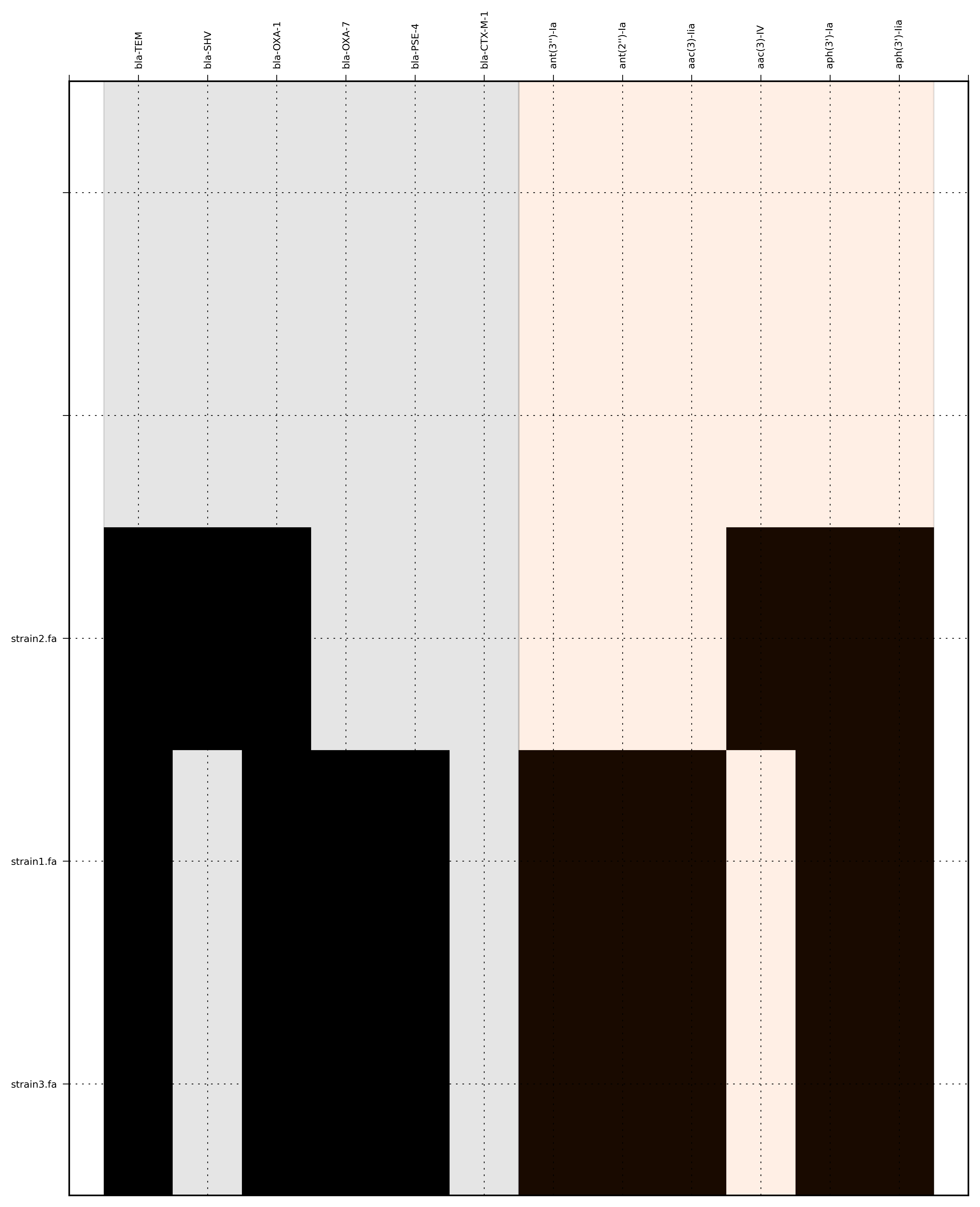
The toy assemblies and consesuses were generated such that:
- strain1 was missing: 70-shv86, 70-ctx143 and 70-aac3(IV)380 with mis-assembly of 70-aphA(1)1310 & 70-tem8674
- strain2 was missing: 70-oxa(7)295, 70-pse(4)348 70-ctx143, 70-aadA1588, 70-aadB1778 and 70-aacC(2)200
- strain2 was missing 70-shv86, 70-ctx143 and 70-aac3(IV)380 with mis-assembly of 70-aphA(1)1310, 70-tem8674 and 70-aadA1588
Run 1 - Looking at only assemblies
Command:
SeqFindR -o run1 -d Antibiotic_markers.fa -a assemblies/ -l

{kind=link}
Run 2 - Combining assembly and mapping consensus data
Command:
SeqFindR -o run2 -d Antibiotic_markers.fa -a assemblies/ -m consensus/ -l

{kind=link}
Run 3 - Combining assembly and mapping consensus data with differentiation between hits
Command:
SeqFindR -o run3 -d Antibiotic_markers.fa -a assemblies/ -m consensus/ -l -r

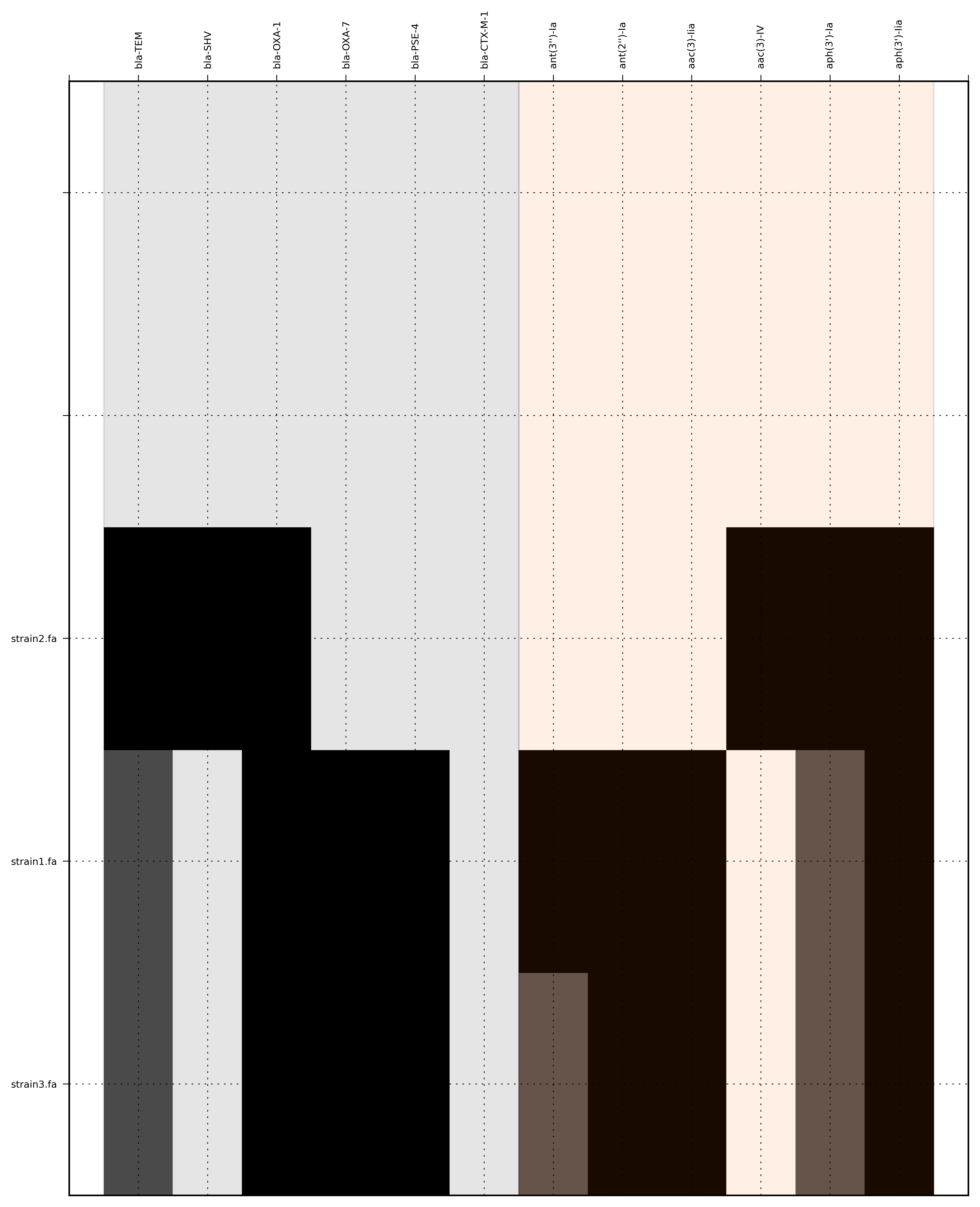{kind=link}
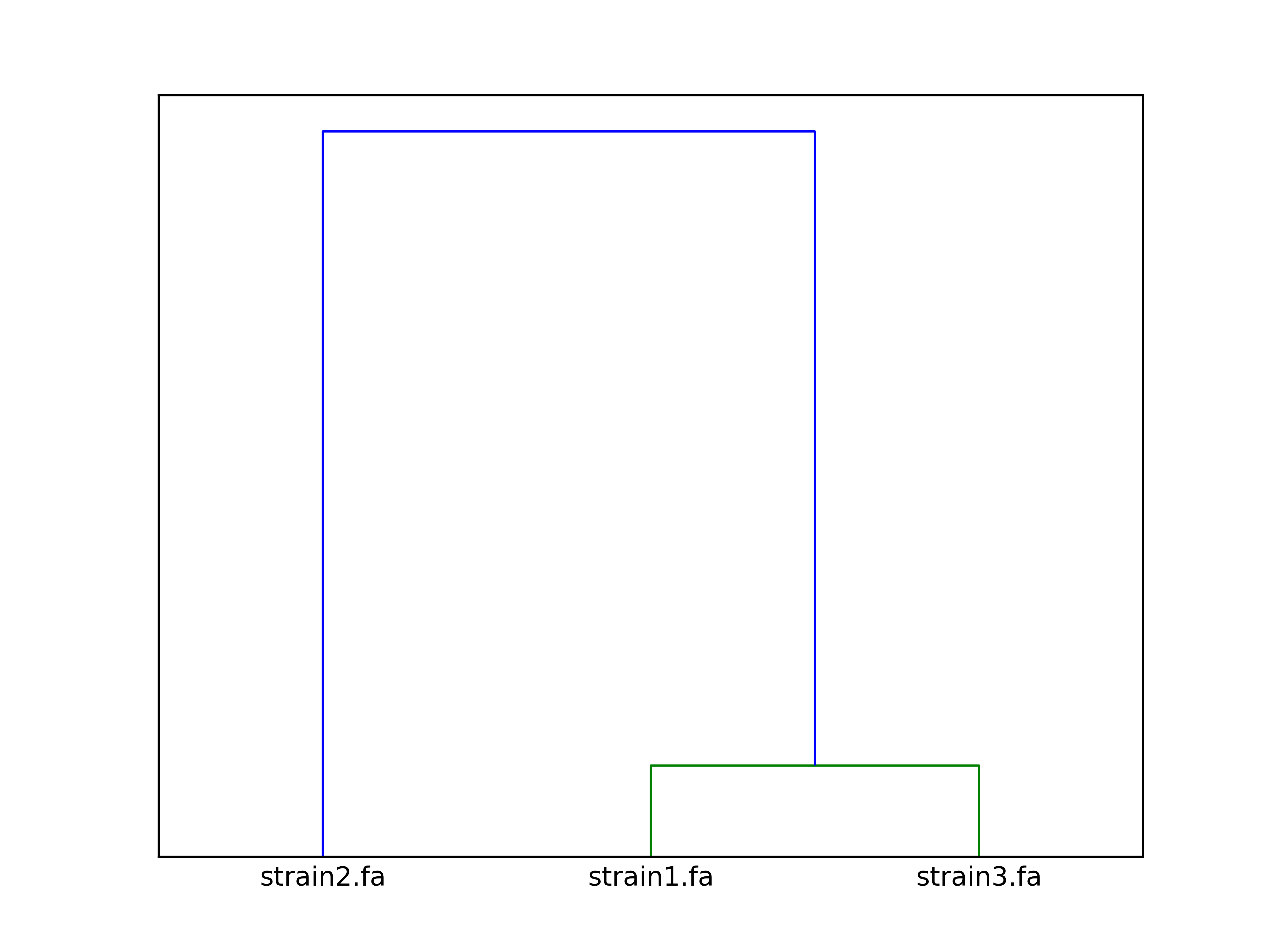
The clustering dendrogram looks like this:

{kind=link}
Run 4 - Combining assembly and mapping consensus data with defined ordering
Command:
SeqFindR -o run4 -d Antibiotic_markers.fa -a assemblies/ -m consensus/ -l -i dummy.order -r

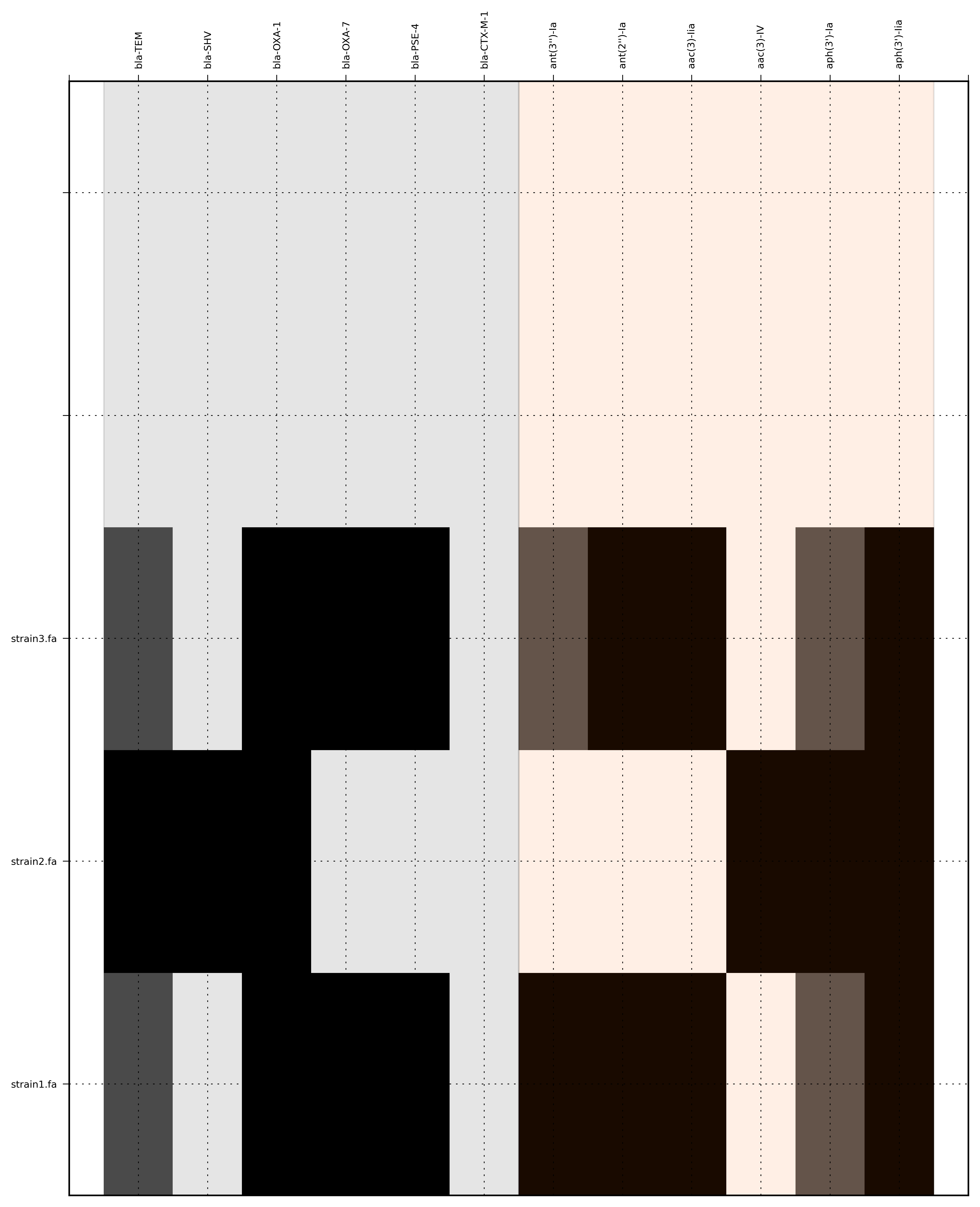{kind=link}
SeqFindR usage options
Help listing:
Usage: SeqFindR.py -o OUTPUT -d DB -a ASS [-h] [-v] [-t TOL] [-m CONS]
[-i INDEX] [-l] [-c COLOR] [-r]
optional arguments:
-h, --help show this help message and exit
-v, --verbose verbose output
-o OUTPUT, --output OUTPUT [Required] output prefix
-d DB, --db DB [Required] full path database fasta file
-a ASS, --ass ASS [Required] full path to dir containing assemblies
-t TOL, --tol TOL Similarity cutoff (default = 0.95)
-m CONS, --cons CONS full path to dir containing consensuses (default = None)
-i INDEX, --index INDEX maintain order of index (no cluster) (default = None)
-l, --label_genes label the x axis (default = False)
-c COLOR, --color COLOR color index (default = None)
-r, --reshape Differentiate between mapping and assembly hits
Licence: ECL by Mitchell Stanton-Cook <m.stantoncook@gmail.com>
Future
- Make into a Web Application
- Trim off first N characters when using mapping consensuses
- More dynamic sizing labeling